

Gaussian GaussView v5.08 Retail
Gaussian GaussView v5.08 Retail | 49.2 Mb
GaussView is the most advanced and powerful graphical interface available for Gaussian. With GaussView, you can import or build the molecular structures that interest you, set up, launch, monitor and control Gaussian calculations, and retrieve and view the results, all without ever leaving the application. GaussView 5 includes many new features designed to make working with large systems of chemical interest convenient and straightforward. It also provides full support for all of the new modeling methods and features in Gaussian 09. This brief introduction will give you a Quick Start to using GaussView 5 to investigate molecules and reactions with Gaussian 09. We invite you to try the techniques described here with your own molecules.
Gaussian GaussView v5.08 Retail
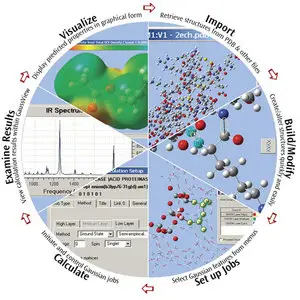
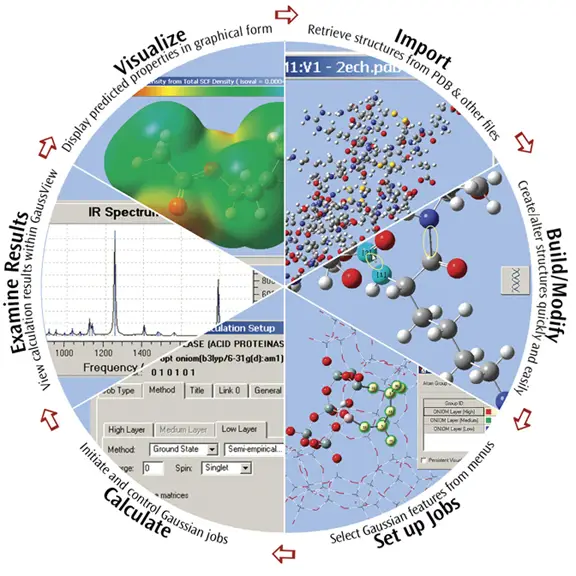
GaussView 5 provides features for every phase of studying large molecular systems, from importing molecules from PDB files, through modifying structural features and setting up ONIOM calculations in Gaussian 09, to viewing and plotting the final results. GaussView can also import many other popular structure exchange formats.
GaussView 5 provides comprehensive support for importing and working with structures from PDB files:
- Select the desired structure(s) from multi-structure files.
- Add hydrogen atoms to all atoms automatically or manually according to user preference.
- Selectively add hydrogen atoms to one or more residues, chains, helices or other defined structural entities.
- Highlight/select atoms in individual residues or secondary structures.
- Quickly determine residue membership for any atom selected with the mouse.
- Easily assign atoms to ONIOM layers based on a variety of flexible criteria.
- Retain residue information within Gaussian 09 calculations and retrieved Gaussian 09 results.
Examine Molecular Structures
- Rotate, translate and zoom in 3D with:
- Mouse operations
- Precision positioning toolbar
- Available in every graphical display - View numeric value for any structural parameter
- Use multiple synchronized or independent views of same structure
- Customize display layout - Manipulate multiple structures individually or as an ensemble
- Display formats: wire frame, tubes, ball & stick/bond type, space fill (CPK) style
- View per-atom labels for element, serial number, NMR shielding (when available)
- Visualize depth with fog feature
- Display stereochemistry info
- Highlight, display or hide atoms based on rich selection capabilities
- Persistent highlighting available
- Convenient palettes:
- Atoms (including hybridization)
- Functional groups
- Rings
- Amino acids (central fragment, amino- or carboxyl-terminated)
- Nucleosides (central fragment, C3’-, C5’-terminated, free forms)
- Custom fragment libraries - Import standard molecule file formats:
- PDB
- Gaussian input, output, checkpoint and cubes files
- Sybyl files: .mol2, .ml2
- MDL files: .mol, .rxn, .sdf
- Crystallographic Information files: .cif
- Optionally include intermediate structures from optimizations etc.
- Multi-structure .sdf and .mol2 files
- Accurately add hydrogens automatically or manually to an entire molecule or to selected residues or secondary structures
- Include/discard waters on PDB import
- Optionally apply standard residue bonding on PDB import
- Include/convert lone pairs for .mol2 - Modify bond type/length, bond angles, dihedral angles
- Rationalize structures with an advanced Clean function
- Recompute bonding on demand
- Constrain structure to specific point group symmetry
- Mirror invert structure
- Invert structure about selected atom
- Place atom/fragment at centroid position of selected atoms
- Define named groups of atoms via:
- Click and marquee selection modes (customizable)
- Complex filters combining atom type, number, MM settings, ONIOM layer
- Select by PDB resuide and/or secondary structure (e.g., helix, chain)
- Expand selections by bond or proximity
- Use groups for display purposes and in Gaussian input - Specify nonstandard isotopes
- Customize fragment placement behavior
- And much much more
Download
No mirrors please
Welcome to my blog!