

BioSolvetIT SeeSAR 14.1.2
BioSolvetIT SeeSAR 14.1.2 | 366.5 mb
BioSolveIT nas released SeeSAR 14.1.2 'Atlas' is an interactive and visual molecular modeling and drug design platform for compound evolution and priorization backed up by science, is intended as an interactive tool for designing/improving ligands for drug discovery.
More polished, more finesse, more efficiency – that’s version 14.1 of SeeSAR ‘Atlas’.
Once again, we’ve rolled up our sleeves to refine and tune the engines and gears of our drug design dashboard SeeSAR. While we were at it, we also put extra effort into new features that will take the user experience to the next level.
In our latest collaboration with YASARA, we are significantly expanding SeeSAR’s molecular modeling feature portfolio with a well-known and popular partner in drug discovery software solutions. The first feature to make its way into SeeSAR’s toolbox is the refinement of structures through energy minimization.
The application areas are particularly diverse: preparing a structure or model (for example from AlphaFold) for molecular modeling studies, docking setups, or entire virtual screening campaigns by eliminating possible torsional conformations and identifying the energetically most favorable arrangement of rotamers.
Additionally, it can be used to refine the poses of docked ligands, eliminating potential issues such as torsions or clashes, or to slightly adjust the size of the binding pocket to uncover new subpockets.
SeeSAR 14.1 ‘Atlas’ — Energy Minimization, Chemical Space Docking, Pharmacophores
Augmentations to Chemical Space Docking
BioSolvetIT SeeSAR 14.1.2
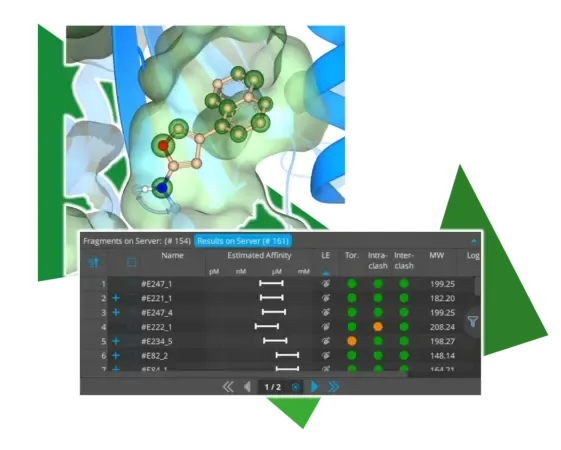
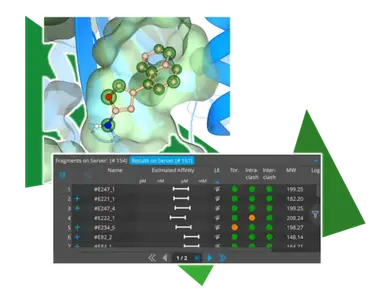
With the introduction of Chemical Space Docking in SeeSAR ‘Atlas,’ we have set the stage for the structure-based exploration of chemical space. To make the workflow even smoother and more efficient, several quality-of-life improvements have been added. One innovation is the visual representation of possible extension vectors for relevant functionalities. As shown in the image above, for example, sp2 hybridized nitrogen atoms now indicate both directions towards which an extension can occur. This visual representation not only reflects the chemically correct steric behavior of the resulting compounds but also emphasizes the growth directions for easier assessment during the assessment of the poses. Furthermore, ligand efficiency (LE) has now been set as the default parameter for sorting Chemical Space Docking results. Especially for the smallest fragments, it come handy to exploit when these exhibit an appropriate LE to enhance the quality of the results. Of course, it is still possible to sort the results by the HYDE score to achieve a balanced mix of both perspectives.
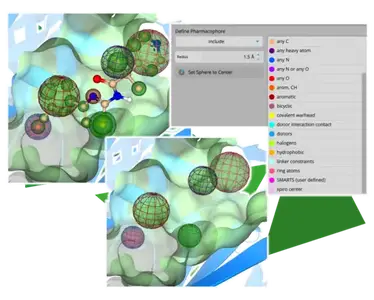
Visual Differentiation of Pharmacophore Constraints
BioSolvetIT SeeSAR 14.1.2
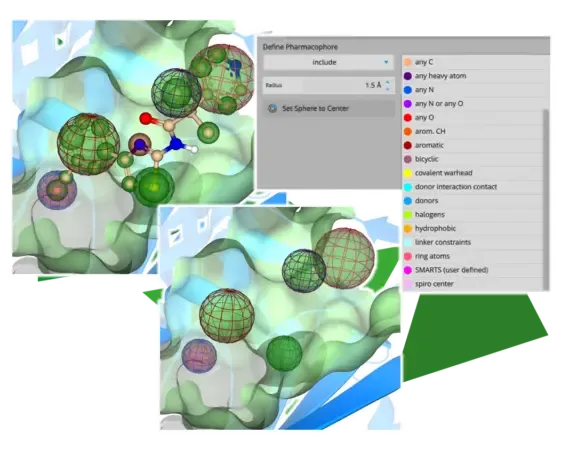
Another highlight is the representation of pharmacophore constraints. The placed spheres now visually reflect the chemical properties of the pharmacophore through color coding and indicate, with green and red coloring, whether they represent inclusive or exclusive filters. Every mode in which constraints may be set (Docking, Space Docking, Inspirator, Analyzer, Similarity Scanner) uniformly supports this feature.
SeeSAR takes its first steps into the world of force-field based molecular mechanics with BioSolveIT entering a strategic partnership with a veteran in the field, YASARA. The first iteration of this integration brings the following enhancements to SeeSAR, powered by the optional YASARA module which comes packaged with the SeeSAR installation.
Key Features
- Energy minimization of proteins and protein-ligand complexes in the Protein Editor mode with 17 force fields to pick from and the AutoSMILES method parameterizing small molecules without the need for manual intervention. The minimized proteins are saved as additional entries to the Protein Editor table.
- The minimization can be done in two levels of flexibility with the user being given the choice of having the backbone fixed and sidechains flexible for minimization, or the entire protein including the backbone to be minimized.
- The minimization can optionally be GPU-accelerated given the availability of a compatible GPU. The usage of the GPU can be explicitly configured via the System Settings in SeeSAR.
- The minimization involves several clean-up steps including the completion of incomplete residues in the structure (absent sequence segments are not covered or completed by this step).
Chemical Space Docking Improvements
- Successful operation of remote docking operations including Space Docking in SeeSAR 14.1 requires upgrading to HPSee 2.1.
- Results returned from server are now sorted by LE by default to avoid the size of fragments biasing the results when sorted by affinity. This change of default is exclusive to the C-S-D mode.
- New visual guidance for the selection of fragments for extension after the anchoring step has been introduced. Whether the extension can happen in either two possible directions or in several possible directions around the linker atom is visually represented by either arrows or circular highlighting in the 3D visualization respectively.
- It is now possible to stop and restart an already running C-S-D step at any point of time so that configuration errors made on the step can be fixed immediately without having to abandon the current workflow and start a new one.
- It is now possible to reconnect to a HPSee server if the connection was lost at some point during execution due to common causes like power failure or loss of connectivity to the network. A "reconnect" button that appears on the interface in such situations can be clicked to attempt reestablishing the connection to the server.
- The interface of the LE and LLE filters in the C-S-D mode has been changed from a button-based interaction to a slider-based interaction to overcome previous limitations in fetching data when non-consecutive combinations of buttons are clicked.
Visualization of Unresolved Segments: Unresolved segments resulting in breaks in protein structures are now visualized in 3D via a yellow/black line connecting the missing parts of the structure.
- Red/black lines indicate unresolved segments that occur within binding sites of proteins indicating the criticality of the missing segments for any downstream workflows such as docking and scoring.
- The visualization can be toggled on/off via a newly added button in the Visualization settings.
Enhanced Pharmacophore Visualization: The display of pharmacophore constraints has been enhanced to facilitate easier color-based visual identification and differentiation.
Each constraint type has been assigned an individual color while the "include" or "exclude" setting is communicated via a green or red highlighting around the constraint sphere.
Miscellaneous and Quality of Life Enhancements
- SeeSAR 14.1 brings significant improvements in cross-platform consistency for docking and scoring workflows.
- A new molecular descriptor/property namely, "Maximum number of consecutive rotatable bonds" has been introduced in SeeSAR to enable sorting and filtering of molecules based on conformational flexibility as implied by this property.
- Molecules from a mode can now be added to other modes like the Docking Mode and Similarity Scanner as "references" and not "templates".
- A molecule added as reference to the Docking Modes can be either used as a "template" to guide the docking when the "template docking" button is clicked, and is considered as a "visual reference" when the "standard docking" button is clicked. This behavior applies to the local, external and the Space Docking Mode.
- All numeric properties that are available for filtering in the Analyzer can now be viewed by switching the respective columns in all modes that have molecule tables to enable sorting of datasets based on these properties.
- Protein-ligand complexes from a mode can be transferred to the Protein Editor either via the context menu on individual entries or by checking multiple entries and adding them to the toolbar via the toolbar button. To edit or minimize one of the proteins added via this mechanism, the entry must be added again to the Protein Editor via the context menu to bring it to the edit state.
- .cif and .mcif files can now be associated with SeeSAR as the default program to open these files by directly double clicking on them, similar to how it is with .pdb files.
- When a SeeSAR project cannot be opened, the user now gets a more informative explanation about the potential reasons (e.g., version incompatibility or file corruption) as to why the project cannot be opened.
Key Features
- Energy minimization of proteins and protein-ligand complexes in the Protein Editor mode with 17 force fields to pick from and the AutoSMILES method parameterizing small molecules without the need for manual intervention. The minimized proteins are saved as additional entries to the Protein Editor table.
- The minimization can be done in two levels of flexibility with the user being given the choice of having the backbone fixed and sidechains flexible for minimization, or the entire protein including the backbone to be minimized.
- The minimization can optionally be GPU-accelerated given the availability of a compatible GPU. The usage of the GPU can be explicitly configured via the System Settings in SeeSAR.
- The minimization involves several clean-up steps including the completion of incomplete residues in the structure (absent sequence segments are not covered or completed by this step).
Chemical Space Docking Improvements
- Successful operation of remote docking operations including Space Docking in SeeSAR 14.1 requires upgrading to HPSee 2.1.
- Results returned from server are now sorted by LE by default to avoid the size of fragments biasing the results when sorted by affinity. This change of default is exclusive to the C-S-D mode.
- New visual guidance for the selection of fragments for extension after the anchoring step has been introduced. Whether the extension can happen in either two possible directions or in several possible directions around the linker atom is visually represented by either arrows or circular highlighting in the 3D visualization respectively.
- It is now possible to stop and restart an already running C-S-D step at any point of time so that configuration errors made on the step can be fixed immediately without having to abandon the current workflow and start a new one.
- It is now possible to reconnect to a HPSee server if the connection was lost at some point during execution due to common causes like power failure or loss of connectivity to the network. A "reconnect" button that appears on the interface in such situations can be clicked to attempt reestablishing the connection to the server.
- The interface of the LE and LLE filters in the C-S-D mode has been changed from a button-based interaction to a slider-based interaction to overcome previous limitations in fetching data when non-consecutive combinations of buttons are clicked.
Visualization of Unresolved Segments: Unresolved segments resulting in breaks in protein structures are now visualized in 3D via a yellow/black line connecting the missing parts of the structure.
- Red/black lines indicate unresolved segments that occur within binding sites of proteins indicating the criticality of the missing segments for any downstream workflows such as docking and scoring.
- The visualization can be toggled on/off via a newly added button in the Visualization settings.
Enhanced Pharmacophore Visualization: The display of pharmacophore constraints has been enhanced to facilitate easier color-based visual identification and differentiation.
Each constraint type has been assigned an individual color while the "include" or "exclude" setting is communicated via a green or red highlighting around the constraint sphere.
Miscellaneous and Quality of Life Enhancements
- SeeSAR 14.1 brings significant improvements in cross-platform consistency for docking and scoring workflows.
- A new molecular descriptor/property namely, "Maximum number of consecutive rotatable bonds" has been introduced in SeeSAR to enable sorting and filtering of molecules based on conformational flexibility as implied by this property.
- Molecules from a mode can now be added to other modes like the Docking Mode and Similarity Scanner as "references" and not "templates".
- A molecule added as reference to the Docking Modes can be either used as a "template" to guide the docking when the "template docking" button is clicked, and is considered as a "visual reference" when the "standard docking" button is clicked. This behavior applies to the local, external and the Space Docking Mode.
- All numeric properties that are available for filtering in the Analyzer can now be viewed by switching the respective columns in all modes that have molecule tables to enable sorting of datasets based on these properties.
- Protein-ligand complexes from a mode can be transferred to the Protein Editor either via the context menu on individual entries or by checking multiple entries and adding them to the toolbar via the toolbar button. To edit or minimize one of the proteins added via this mechanism, the entry must be added again to the Protein Editor via the context menu to bring it to the edit state.
- .cif and .mcif files can now be associated with SeeSAR as the default program to open these files by directly double clicking on them, similar to how it is with .pdb files.
- When a SeeSAR project cannot be opened, the user now gets a more informative explanation about the potential reasons (e.g., version incompatibility or file corruption) as to why the project cannot be opened.
BioSolvetIT SeeSAR 14.1.2
SeeSAR is your intuitive, visual drug design platform. Converting every step of your drug discovery process from virtual screening to fragment-based design. SeeSAR fosters ideation in the most fun and comprehensive way. SeeSAR functionalities: docking simulation, scaffold replacement, fragment growing, affinity evaluation, molecular surface, torsion quality evaluation, small molecules superposition, ligand property and ADME property calculation etc.
Introduction to SeeSAR: Drug Discovery with ‘Midas’
In this BioSolveIT workshop, we embark on a journey into the world of the drug discovery dashboard SeeSAR: A cutting-edge tool that has undergone a remarkable power gain over the years. During this workshop, we will offer a comprehensive introduction to its various operational “modes”. Modes, pleasingly designed, cover a wide array of tasks such as molecule editing, docking, and target-ligand complex assessment. Additionally to the structure-based methods, we will discuss how SeeSAR can facilitate ligand-based drug discovery (LBDD).
BioSolveIT GmbH is a Bio- and Cheminformatics company. Its core businesses are software, services, and research collaborations. With three founders in academia, BioSolveIT has its backbone in research and catalyzes the genesis of products off of basic research successes. Best known is its molecular docking software FlexX. With the FTrees program BioSolveIT took leadership in ultra-fast virtual high throughput screening using a fragment reassembly based approach.
Owner: BioSolveIT GmbH
Product Name: SeeSAR 'Atlas'
Version: 14.1.2
Supported Architectures: x64
Website Home Page : www.biosolveit.de
Languages Supported: english
System Requirements: Windows *
Size: 366.5 mb
BioSolvetIT SeeSAR 14.1.2
Please visit my blog
Added by 3% of the overall size of the archive of information for the restoration
No mirrors please

![BioSolvetIT SeeSAR 14.1.2]()
Added by 3% of the overall size of the archive of information for the restoration
No mirrors please
BioSolvetIT SeeSAR 14.1.2
